Overview and Legislative Timeline
The EU Pharma Package represents the most comprehensive reform of EU pharmaceutical legislation in over 20 years. It consists of a new Regulation and a new Directive, which will replace the current framework (Directive 2001/83/EC and Regulation (EC) No 726/2004).
Originally proposed by the European Commission in April 2023, the package was subject to extensive negotiations between the European Parliament and the Council throughout 2024–2025. A political agreement was reached on 11 December 2025, and the agreed outcome was subsequently reflected in the compromise texts published on 6 March 2026. Formal adoption is expected imminently, but the text is now not expected to change.
Once formally adopted, both the Regulation and the Directive will enter into force on the twentieth day following their publication in the Official Journal of the European Union. The Regulation will become applicable 24 months after its entry into force, while Member States will have 24 months from the Directive’s entry into force to transpose it into national law.
The pharma reform is extensive and touches on a number of topics, but we tend to see queries from pharma clients around changes in the following areas.
Data and Marketing Protection - Changes from the Current 8+2(+1) Regime
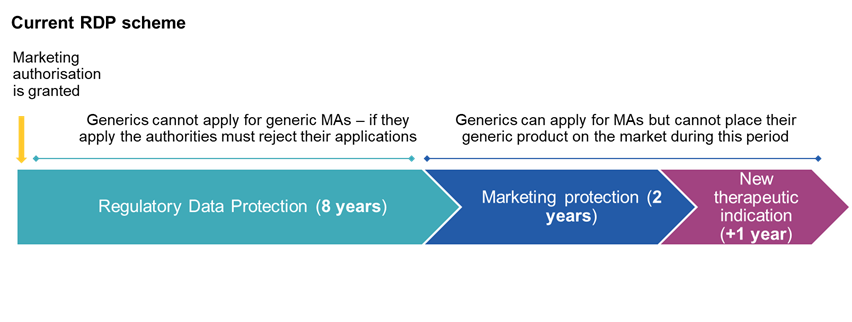
The current regulatory data protection (RDP) regime provides for: 8 years of data protection during which Gx/Bx cannot rely on the data of the reference product to obtain MAs, and 2 years of marketing protection during which MAs can be obtained but the product cannot be placed on the market. These 2 years can in some circumstances be extended by a further year (e.g. if approval is obtained for a new therapeutic indication).
In the various drafts of the new pharma reform, different durations were proposed for the above periods, mostly aimed at shortening the overall RDP period for originators, which have thankfully been reversed in the final version of the legislation. The settled position is now as follows:
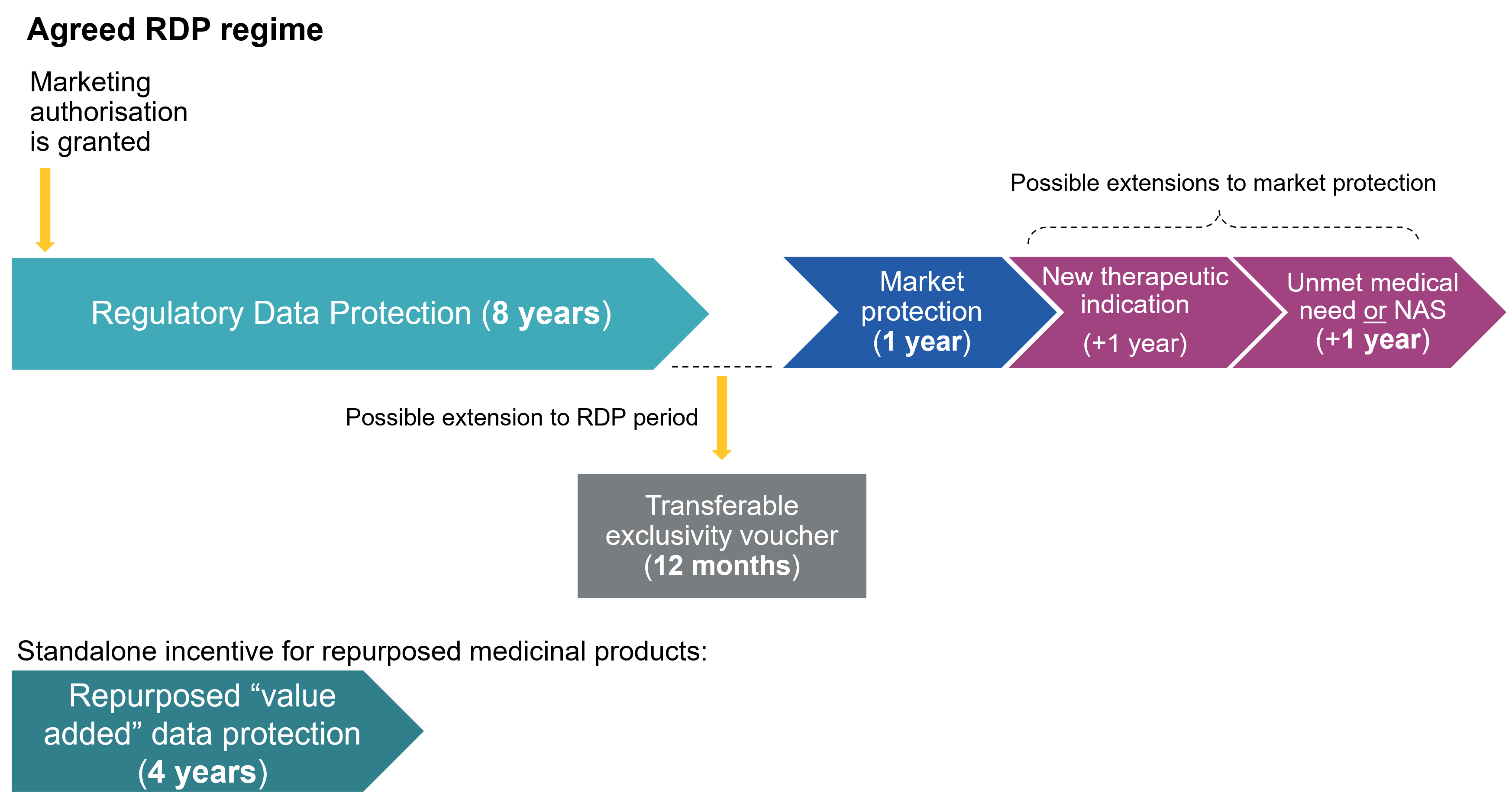
- Data protection. The standard period of regulatory data protection remains 8 years. Companies may also obtain a 'Transferable Exclusivity Voucher' (TEV) for obtaining approval of a priority antimicrobial – see further below. This voucher can be used to extend the data protection of any centrally approved product (including the antimicrobial itself) that the company markets by a total of 12 months, bringing the total data protection period to 9 years.
- Market protection. The reform introduces a restructured system of market protection, moving away from the current “8+2(+1)” model towards a more articulated 8+1(+1)(+1) framework. Under the proposed Directive, the baseline period of market protection is reduced from two years to one year, departing from the current regime. It is possible to obtain additional one-year extensions, subject to the fulfilment of specific conditions.
- Extensions. The final text retains a limited set of extensions, reflecting a negotiated balance between the different institutional positions.
- A one-year extension of market protection may be granted for the approval of a new therapeutic indication demonstrating a significant clinical benefit over existing therapies (similar to the current +1 regime). This extension can be awarded only once, in line with the current framework.
- This may be combined with one further one-year extension, available in two alternative scenarios:
- where the medicinal product addresses an unmet medical need, as defined in the proposed Directive; or
- where the medicinal product contains a new active substance (NAS) and any of the following criteria apply:
- clinical trials use an evidence-based comparator in line with the European Medicine Agency’s (EMA) scientific advice and the MA application was first filed in the EU (or within 90 days of the first non-EU filing); or
- the clinical trials use an evidence-based comparator in line with EMA’s scientific advice and efficacy clinical trials were conducted in more than one Member State; or
- efficacy clinical trials were conducted in more than one Member State and the MA application was first filed in the EU (or within 90 days of the first non-EU filing) — only possible where the MA applicant can justify that a comparator trial (i.e., scenario (i) or (ii)) is not feasible.
- This may be combined with one further one-year extension, available in two alternative scenarios:
- A one-year extension of market protection may be granted for the approval of a new therapeutic indication demonstrating a significant clinical benefit over existing therapies (similar to the current +1 regime). This extension can be awarded only once, in line with the current framework.
- Repurposed (value-added) medicinal products. The final text confirms the introduction of a standalone incentive in the form of a four-year period of data protection for repurposed (or “value-added”) medicinal products. This applies where a new therapeutic indication is authorised for a known active substance, provided that the indication has not previously been authorised in the Union, and subject to compliance with specific conditions set out in the proposed Directive (including requirements as to evidence generation and regulatory submission).
In conclusion, originators may still benefit from up to 11 years of protection (and in exceptional cases with the TEV, 12 years). However, access to the full duration will increasingly depend on meeting defined clinical and public health objectives. In this sense, the revised RDP framework does not drastically reduce the quantitative level of protection, but rather reconfigures its allocation, aligning regulatory incentives more closely with clinical value, innovation priorities and timely patient access across the EU.
Orphan Medicinal Products
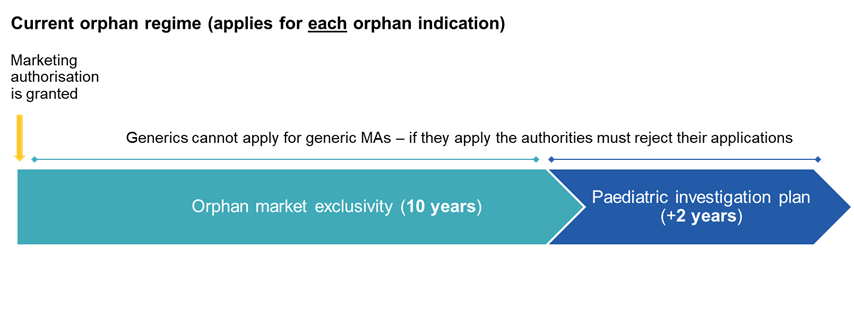
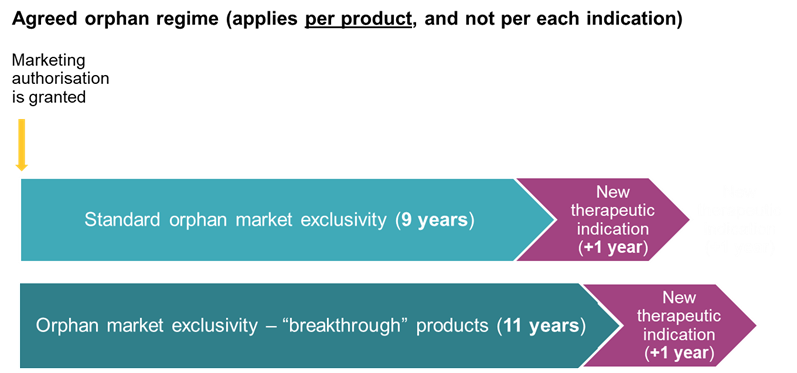
The framework for orphan medicinal products is significantly restructured, moving from an indication-based system, under which each orphan indication could benefit from a separate 10-year period of orphan market exclusivity (OME), to a product-based approach, whereby exclusivity is anchored to the first orphan marketing authorisation granted in the Union, irrespective of subsequent orphan indications for the same active substance.
Furthermore, the standard period of OME is reduced from 10 years to 9 years. On the flipside, an extended period of 11 years is available for so-called “breakthrough” orphan medicinal products, i.e. those intended for conditions for which no satisfactory treatment exists and which provide a clinically relevant reduction in morbidity or mortality. Both the 9-year and 11-year periods may be extended by an additional 12 months where, at least two years before expiry, the marketing authorisation holder obtains approval for a new therapeutic indication covering a different orphan condition.
Orphan market exclusivity (OME) continues to provide a broader form of protection, in that it prevents the approval (the way data protection does) of all similar medicinal products, including innovative competitors, and not only generics or biosimilars.
Additionally, companies developing promising orphan medicinal products may benefit from earlier and more structured regulatory support, including scientific guidance at an earlier stage of development, with a view to accelerating patient access.
For paediatric incentives affecting orphan medicines, see below.
Boost to Antibiotics
Addressing antimicrobial resistance (AMR) is a key objective of the EU Pharma Package, which introduces a novel incentive in the form of a transferable exclusivity voucher (TEV).
Developers of a priority antimicrobial - i.e. a product targeting multi-drug resistant organisms and demonstrating a significant clinical benefit in relation to AMR may be granted a TEV, entitling the holder to one additional year of regulatory data protection. This may be applied either to the antimicrobial itself or to another authorised medicinal product, and may also be transferred (sold) to third parties, thereby creating a tradable incentive.
The TEV is granted by the European Commission, following a scientific assessment by the EMA, and is valid for five years. Its award is subject to strict conditions, including ensuring adequate supply within the EU, disclosure of financial support in the development of the antimicrobial, and timely EU filing requirements.
The use of the voucher is also subject to limitations: it may only be applied during the fifth or sixth year of the data protection period (to avoid last-minute extensions), and cannot be used for products exceeding €490 million in annual EU sales in any of their first four years (the so-called “anti-blockbuster” cap).
Paediatric Medicinal Products
As regard paediatric medicinal product, following completion of an agreed paediatric investigation plan (PIP), and where a medicinal product is authorised for a paediatric indication, the marketing authorisation holder (MAH) is required to place the product on the market with that paediatric indication within two years in the relevant Member States.
In parallel, the reform strengthens safeguards to ensure continuity of supply for paediatric medicines. Where an MAH intends to withdraw a medicinal product authorised for a paediatric indication from the market, it must provide at least 12 months’ prior notice to the relevant competent authorities. The MAH must also take appropriate measures to ensure continued patient access, including, where necessary, by transferring the marketing authorisation to a third party or by allowing a third party to rely on the product dossier.
As regards its interplay with patent-based incentives, the reform removes the current two-year extension of orphan exclusivity linked to the completion of a paediatric investigation plan. Instead, the six-month extension of the supplementary protection certificate (SPC) becomes available to orphan medicines, thereby aligning the incentive framework with that applicable to non-orphan medicinal products.
Changes to MA Applications and Processes
The reform introduces a number of targeted changes to marketing authorisation applications and related regulatory processes, aimed at increasing efficiency and strengthening environmental oversight, while at the same time reinforcing the resilience of the EU medicines supply chain.
The Reform shortens the scientific assessment timeline for centralised MA applications. The EMA will generally have 180 days to issue its opinion, reduced from the current 210-day timetable, with a further reduction to 150 days for medicinal products of major interest for public health.
Further, the reform reinforces environmental risk assessment (ERA) requirements. All MA applications must include an ERA addressing the environmental impact of the medicinal product and its components, including whether they exhibit characteristics such as persistence, bioaccumulation, or toxicity. Enhanced requirements apply in particular to antimicrobials and to medicinal products containing or consisting of GMOs, which remain subject to a dedicated assessment framework.
Supply Shortages
In parallel, the reform introduces a significantly strengthened framework on supply obligations and shortages management, combining monitoring, prevention and enforcement mechanisms:
- Marketing authorisation holders of originator products are subject to enhanced supply obligations, including the requirement to ensure that medicinal products are made available in a Member State when requested. This includes, where applicable, engagement with pricing and reimbursement procedures, participation in procurement processes and implementation of supply roll-out plans. Failure to ensure continuous supply over a sustained period may result in loss of regulatory protection and market exclusivity in the relevant Member State, thereby enabling earlier market entry by generic/biosimilar manufacturers.
- The framework also introduces mandatory shortage prevention plans. MAHs of prescription medicinal products, as well as those products identified as critical by the Commission, must establish and maintain such plans, based on regular risk assessments of the supply chain and appropriate mitigation measures.
In addition, notification obligations are strengthened. MAHs must inform the competent authorities of any temporary or permanent supply disruption. Where a disruption is expected to last for an extended period (e.g. two years or more), advance notice - typically at least six months -must be provided, enabling authorities to anticipate and manage potential shortages.
At EU level, the Commission will maintain a Union list of critical medicinal products, based on input from national competent authorities, and vulnerabilities in the supply chains of those products will be assessed and issue recommendations issued to enhance security of supply.
Bolar Exemption
The Bolar exemption permits generic and biosimilar companies to carry out certain preparatory activities in view of market entry without infringing patents or supplementary protection certificates (SPCs).
Under the current framework, the wording of the exemption has given rise to somewhat differing interpretations across Member States, resulting in a fragmented body of case law.
The final reform text aims to harmonise the position across the EU, but in doing so also significantly expands the scope of the Bolar exemption. In particular, it states that the exemption now covers not only activities strictly necessary for obtaining a marketing authorisation, but also pricing and reimbursement procedures, health technology assessment (HTA) processes, and - importantly - participation in public procurement procedures (ie tenders), provided that any resulting supply takes place only after expiry of the relevant protection. The exemption is also explicitly extended to cover activities carried out by third parties, including API manufacturers and other supply chain operators.
This is a significant broadening of the Bolar exemption, as it now covers various acts that have traditionally been considered to be infringing (e.g. 'offering for sale' in a tender), or imminently infringing for the purposes of seeking a preliminary injunction. The new text of the Reform now expressly states that decisions of public authorities in relation to any of the above mentioned activities shall not be considered as infringing intellectual property rights, and that intellectual property rights of the reference medicinal product shall not represent a valid ground to refuse, revoke or suspend such decisions
Next Steps
As noted above, formal adoption of the EU Pharma Package is expected imminently. The new Regulation and Directive will enter into force on the twentieth day following publication in the Official Journal, with the Regulation becoming applicable after 24 months and Member States having an equivalent period to transpose the Directive. The revised framework is therefore expected to become fully operational around 2028, at least for the main provisions.
In relation to regulatory exclusivity specific transitional provisions will apply. Marketing authorisations already granted, as well as applications submitted before the date of application of the new regime, will continue to be governed by the current data and market protection rules, thereby ensuring continuity for products already in development.
Key contacts

Priyanka Madan
Senior Associate, London and Milan

Martina Maffei
Of Counsel, Milan

Hannah Marsh
Associate, London
Disclaimer
The articles published on this website, current at the dates of publication set out above, are for reference purposes only. They do not constitute legal advice and should not be relied upon as such. Specific legal advice about your specific circumstances should always be sought separately before taking any action.